
A simple initial example is running MD simulations of an RNA duplex that contains a pseudo-uridine (PSU) modification. We will base this example on the Nuclear Magnetic Resonance (NMR) determined structure deposited with PDB ID 6I1W [14] that contains the PSU residue at position 5 of the sense strand. After downloading the legacy PDB Format file (6I1W.pdb) some cleaning is required to have a working PDB file that we can then use for building parameter and input coordinate files for MD simulations. There are multiple ways to do this. In this case, we will use the prepareforleap command available in CPPTRAJ [15]. Briefly, this command removes information that the PDB file includes that is either not needed by or would cause issues with LEaP, removes water molecules, and performs a brief check of the residues to report problems that LEaP might encounter. In a text file (prepareforleap.cpptraj), add the commands
parm 6I1W.pdb
loadcrd 6I1W.pdb name edit
prepareforleap crdset edit name FromPrepForLeap \
out FromPrepForLeap−tleap.in leapunitname x \
pdbout FromPrepForLeap.pdb nowat noh
gowe execute the command using:
$ cpptraj -i prepareforleap.cpptraj
CPPTRAJ will open the 6I1W.pdb file, delete the water molecules, strip hydrogens and generate a new PDB file (from-prepareforleap.pdb). The CPPTRAJ output will print a warning referring to the PSU residue:
Potential problem : PSU_5_A is an unrecognized name and may not have parameters .
This is because PSU is not a residue with parameters in the standard AMBER force fields. We will now use modXNA to create the parameters for the PSU residue. Even though modXNA is available in AmberTools25, it is recommended that the user download the latest version of modXNA from the website (https://modxna.chpc.utah.edu/ or this article’s github repository (https://github.com/ManghraniA/ModXNALiveComms). ModXNA requires an input file that declares the backbone, the sugar, and the base. Looking at the catalog of available fragments on the modXNA website, we need the RNA phosphate backbone (modXNA code RPO), a ribose sugar (modXNA code RC3), and the pseudo uridine base (modXNA code PUU). The input file (PSU.in.modxna, Listing 2) is:
##modXNA input file
RPO RC3 PUUWe run the script with:
$ modXNA.sh -i PSU.in.modxna -m PSU
The -i flag indicates the input file, and the -m flag indicates the name that we want for the generated nucleotide. Without the -m flag, modXNA will generate a random 3-letter residue name. The script will generate multiple temporary files (with the prefix “tmp.”) that are useful to troubleshoot if something went wrong. If the script is completed successfully, we will have a PSU.lib file with the nucleotide, created from the selected fragments in the modXNA input script. We can check the structure of the generated nucleotide using the tmp.opt.pdb file in any molecular visualizer:
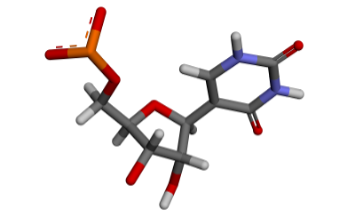
We can now build our Amber topology and coordinates files using LEaP. We use the following tleap script (build.tleap):
###########################
source leaprc.DNA.OL15
source leaprc.RNA.OL3
source leaprc.water.opc
##########################
## Load the LIB file created with modXNA
loadoff PSU.lib
## Load the force mod file included in modXNA
loadamberparams /home/user/modXNA/dat/frcmod.modxna
######################################
x = loadpdb FromPrepForLeap.pdb
solvateoct x OPCBOX 9.0
addionsrand x Na+ 0
addionsrand x Cl− 0
saveamberparm x complex.parm7 complex.crd
charge x
quitWe can run the above script with the following command:
$ tleap -s -f build.tleap
LEaP will load the declared AMBER force fields, load the LIB file and the frcmod files, then load the PDB structure generated from the prepareforleap command. The files complex.parm7 and complex.crd will be successfully generated.
*Note that atom names for residues built by modXNA may not match your particular PDB file. In this case, it is usually acceptable to keep commonly named atoms, delete atoms with naming differences, and have LEaP build in the remainder of the nucleotide based on the coordinates and connectivity in the lib file.