
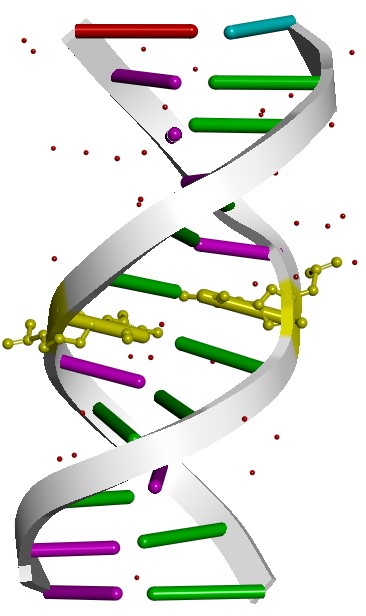
In this example we are going to use modXNA to generate a methyl-cytosine residue to model the structure 329D from the Protein Databank.
The structure consists of two 12-mer strands of DNA with the modified 5CM residue in position 6 and 18 (highlighted in yellow):
We can use modXNA to create our methylated cytosine residue.
Download the latest version of modXNA and extract the file:
tar xvfz modXNA-<latest-version>.tar.gz
modXNA requires an input file (text file) that indicates what fragments to use to create a nucleotide. The text file has the form:
<backbone ID> <sugar ID> <base ID>
For our example, we need:
- the DNA backbone, which is fragment code: DPO
- the DNA sugar, which is fragment code: DC2
- and the methylated cytosine, which is fragment code: M5C
Check the codes from the fragment catalog. Create a text file (for example: in.modxna) that will look like:
### Commented line
### DNA residue with methyl-C
DPO DC2 M5CRun modXNA using:
$ ./modxna.sh -i in.modxna
The program will look for the fragments in the dat/ directory, remove capping groups, join the fragments together, adjust the charge and run a small minimization to optimize the resulting structure.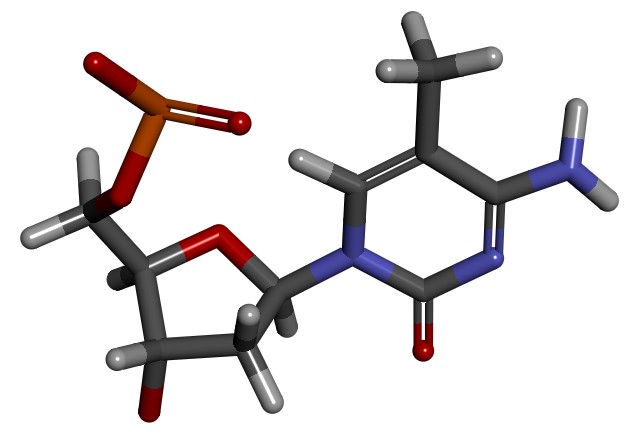
When modXNA is run, it generates a random name for our new residue. In our example, the name is: RSS – modXNA generates an RSS.lib file that we can use to build an AMBER topology and coordinate file to run MD simulations.
To incorporate our new residue into our PDB file, we can change the residue name from the PDB (which is 5CM) to RSS (the name of our lib file) and save the PDB file with a new name (i. e. 329d-rss.pdb).
## Amber OL15 force field is used to parameterize
## canonical nucleotides
source leaprc.DNA.OL15
source leaprc.water.opc
## Load modxna frcmod & library files - the random
## three-letter code ("RSS") generated from modxna MUST ## match the residue name in the structure PDB
loadamberparams modxna/dat/frcmod.modxna
loadoff RSS.lib
## Load and solvate the system
dna = loadpdb 329d-rss.pdb
addions dna Na+ 0
addions rna Cl- 0
solvateoct dna OPCBOX 10.0
## Add additional ions for appropriate environment, in ## this case our volume is 182273.67 A^3 and we would
## like a 200 mM NaCl concentration
addions dna Na+ 22
addions dna Cl- 22
saveamberparm dna dna.topo dna.coordsWhen we run the LeAP program, we will see that there are two errors:
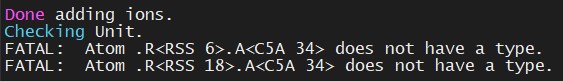
This is because the fragment that we used for methy-cytosine does not have an atom with the name C5A – we can delete the lines that have that atom from the PDB input file and let LeAP fill in the missing atoms based on our generated RSS.lib file.
Once we have deleted the lines that contain the C5A atom, we can safely re-run LeAP and the topology and coordinate file will be generated that we can use in a molecular dynamics simulation.